
TGFβ is a cytokine with many, often paradoxical, roles in cancer biology. Acting as a tumor suppressor, TGFβ exerts negative control on epithelial cell proliferation - a function that tumor cells must overcome in order to progress to malignant disease. On the other hand, TGFβ also acts as a pro-tumorigenic factor, promoting malignant phenotypes such as invasion, and exerting pro-tumor effects on components of the tumor microenvironment, such as suppression of anti-tumor immune responses. The pleiotropic role of this cytokine in tumorigenesis and cancer progression complicates the potential targeting of its signaling pathway for cancer therapy. Members of the Paulovich Laboratory, in the Fred Hutch Clinical Research Division, joined a multi-institute collaborative effort to investigate the poorly understood role of TGFβ in DNA damage repair and genomic instability. Their work, recently published in Science Translational Medicine, uncovered an unexpected reciprocal relationship between TGFβ signaling and error-prone DNA damage repair in cancer, ultimately leading to implications for therapeutic outcomes.
Though a connection between TGFβ and genomic instability has been recognized for decades, the biology underlying this phenomenon has yet to be fully elucidated. More recently, it has become appreciated that TGFβ signaling regulates key proteins that coordinate the DNA damage response. Two classical DNA repair pathways are homologous recombination repair (HR), which utilizes a second, intact DNA template to guide repair, and non-homologous end-joining (NHEJ), in which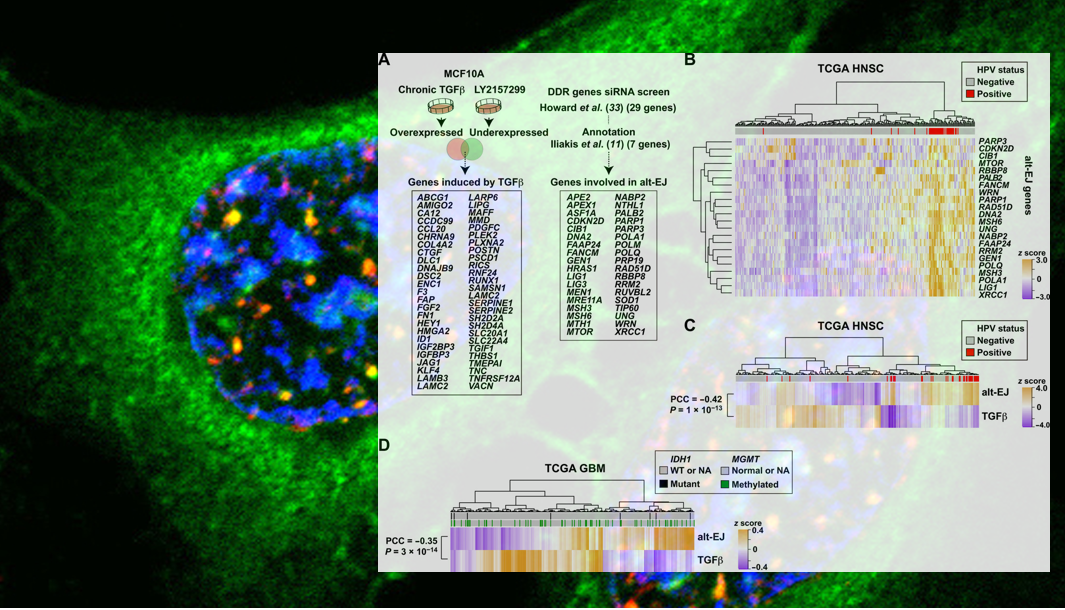 broken DNA strands are directly ligated without the need for a template. When these classical repair pathways are inoperable, a third, non-classical repair mechanism called alternative end-joining (alt-EJ) is thought to kick in as a last resort, though this pathway is highly error-prone, causing frequent genomic deletions and insertions at the repair sites. Importantly, TGFβ signaling reinforces classical DNA damage response pathways, and results in lower levels of alt-EJ. Understanding the mechanisms underlying DNA repair decisions and defects in cancer cells can provide therapeutic opportunities for exploiting disease vulnerabilities. Thus, the Paulovich Lab joined with a team of collaborators led by Dr. Mary Helen Barcellos-Hoff at USCF and Dr. Miquel Angel Pujana at Institut Català d'Oncologia (ICO) in Barcelona to elucidate the mechanistic relationships between TGFβ signaling, DNA damage, and genomic instability. “This collaboration pins down TGFβ’s role in regulating DNA repair pathway, which is perhaps the least well-understood but may finally give a handle how it should be targeted to improve cancer outcomes” Dr. Barcellos-Hoff said.
broken DNA strands are directly ligated without the need for a template. When these classical repair pathways are inoperable, a third, non-classical repair mechanism called alternative end-joining (alt-EJ) is thought to kick in as a last resort, though this pathway is highly error-prone, causing frequent genomic deletions and insertions at the repair sites. Importantly, TGFβ signaling reinforces classical DNA damage response pathways, and results in lower levels of alt-EJ. Understanding the mechanisms underlying DNA repair decisions and defects in cancer cells can provide therapeutic opportunities for exploiting disease vulnerabilities. Thus, the Paulovich Lab joined with a team of collaborators led by Dr. Mary Helen Barcellos-Hoff at USCF and Dr. Miquel Angel Pujana at Institut Català d'Oncologia (ICO) in Barcelona to elucidate the mechanistic relationships between TGFβ signaling, DNA damage, and genomic instability. “This collaboration pins down TGFβ’s role in regulating DNA repair pathway, which is perhaps the least well-understood but may finally give a handle how it should be targeted to improve cancer outcomes” Dr. Barcellos-Hoff said.
The Paulovich lab applied a series of phosphoproteomics and genomics analyses to tumor cells exposed to DNA damage in the presence or absence of an inhibitor of TGFβ signaling. It was previously known that blocking TGFβ signaling leads to decreased activation of ATM, a protein kinase that functions as a central DNA damage response regulator. Leveraging their expertise in targeted mass spectrometry, the researchers identified a swath of DNA damage-induced phosphorylation events that are blocked by TGFβ inhibition, including that of the ATM target BRCA1, an important component of the HR pathway. Analysis of gene expression changes upon TGFβ signaling or blockade revealed induction of HR- and NHEJ-associated transcripts, and suppression of alt-EJ-associated transcripts by TGFβ. To study whether the role of TGFβ in suppressing alt-EJ is linked to its known roles in promoting classical repair mechanisms, the investigators measured repair pathway usage in the presence or absence of TGFβ signaling blockade in combination with molecular HR and NHEJ inducers and inhibitors. Surprisingly, TGFβ suppression continued to induce alt-EJ regardless of classical pathway modulation. Contrary to what was previously known, these findings indicate a distinct mechanism for TGFβ in suppressing alt-EJ, separate from its effects on classical repair pathway mediators.
To examine the interaction between TGFβ signaling and alt-EJ more broadly, the group turned to The Cancer Genome Atlas, a publicly-available database of clinical cancer genomics data. They curated an alt-EJ gene signature for evaluation alongside an existing TGFβ signature across patient samples. Excitingly, the inverse correlation between TGFβ and alt-EJ signatures was broadly present across human solid tumors, consistent with the biology that they had delineated in their own laboratories. Hypothesizing that high alt-EJ would lead to genomic instability, due to the error-prone nature of this repair mechanism, the group analyzed the relationship between tumor genome alterations and TGFβ /alt-EJ signatures. Indeed, tumors that exhibited high alt-EJ and low TGFβ showed higher somatic mutational burdens than those exhibiting the opposite signature pattern, and the mutation types observed were consistent with those expected to be generated from alt-EJ repair. Finally, investigating the prediction that genomic instability would lead to increased susceptibility to genotoxic therapies, they observed better patient outcomes in high alt-EJ, low TGFβ cancers across tumor types in which genotoxic therapies are the standard of care.
“We were thrilled to be a part of this collaborative study using our proteomic platform to help verify an unexpected reciprocal relationship between TGFβ signaling and alt-EJ–mediated DNA repair,” said Dr. Paulovich. “The study shows that, regardless of cancer type, tumors classified as low TGFβ and high alt-EJ are characterized by an insertion-deletion mutation signature containing short microhomologies and are more sensitive to genotoxic therapy.” Looking forward, Dr. Paulovich and collaborators are motivated by the potential implications of these findings for cancer patients. “We are interested in determining if these results can be used to improve the care of cancer patients through a personalized oncology approach, by matching patients to individualized therapies that exploit this vulnerability.”
Article: by Fred Hutchinson Cancer Research Center
Source: Loss of TGFβ signaling increases alternative end-joining DNA repair that sensitizes to genotoxic therapies across cancer types. Sci Transl Med. 2021 Feb 10;13(580):eabc4465. doi: 10.1126/scitranslmed.abc4465. PMID: 33568520.