
The limited availability of patient tissue samples poses a constant challenge for omics researchers, particularly in determining which analyses are feasible based on sample input requirements. Existing parallel workflows for multi-omic analyses have yielded valuable insights but are often restricted to the analysis of one or two post-translational modifications. The MONTE workflow, described by the Carr lab in a recent study published in Nature Communications, addresses these limitations. By leveraging serial HLA-II and HLA-I immunopeptidomics alongside ubiquitylome, proteome, phosphoproteome, and acetylome data, the workflow enables multiple layers of information to be obtained from each sample starting with as little as 25 mg wet weight of tissue.
In order to optimize the workflow, four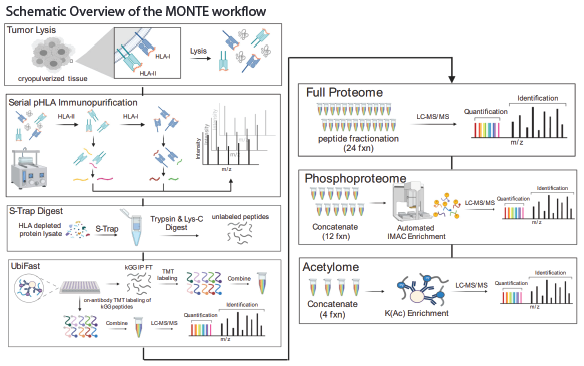 key changes have been made to previously reported protocols: 1.) Samples are first serially enriched for HLA-II and HLA-I immunopeptidomics before downstream multi-omic analyses. This involves the use of specific protease inhibitors tailored to each proteome and PTM-ome, as well as the selection of a pan anti–HLA-DR, -DP, and -DQ antibody mixture. 2.) The traditional 8 M urea cell lysis step was replaced with SDS denaturation and digestion on an S-Trap. This modification facilitated the removal of detergents present in the native lysis buffer used for HLA immunoprecipitation, ensuring cleaner and more accurate results. 3.) An optimized semi-automated, 96-well plate–based workflow was implemented 4.) The flow-through from the HLA enrichment steps was then digested, enriched for ubiquitylated peptides using the UbiFast protocol (ref) prior to collection of proteomic data and enrichments of phosphoryalated and acetylated peptide.
key changes have been made to previously reported protocols: 1.) Samples are first serially enriched for HLA-II and HLA-I immunopeptidomics before downstream multi-omic analyses. This involves the use of specific protease inhibitors tailored to each proteome and PTM-ome, as well as the selection of a pan anti–HLA-DR, -DP, and -DQ antibody mixture. 2.) The traditional 8 M urea cell lysis step was replaced with SDS denaturation and digestion on an S-Trap. This modification facilitated the removal of detergents present in the native lysis buffer used for HLA immunoprecipitation, ensuring cleaner and more accurate results. 3.) An optimized semi-automated, 96-well plate–based workflow was implemented 4.) The flow-through from the HLA enrichment steps was then digested, enriched for ubiquitylated peptides using the UbiFast protocol (ref) prior to collection of proteomic data and enrichments of phosphoryalated and acetylated peptide.
After working to optimize the workflow, the researchers demonstrated the effectiveness of MONTE in a proof-of-concept study on primary patient lung adenocarcinoma (LUAD) tumors. The results from the characterization of LUAD data showed that the MONTE workflow was able to identify several novel HLA-binding peptides, including neoantigens, as well as a number of proteins that were differentially expressed in the tumor samples. Historically, the detection of neoantigens required enrichment from either billions of cells or gram levels of tissue, as neoantigens can represent only 0.01% of all unique peptide identifications. These findings underscore the capability of acquiring additional data layers without compromising data quality, highlighting the power and versatility of the MONTE workflow. Furthermore, the workflow offers insights into tumor immune cell infiltration status and dysregulation of signaling, degradation, and epigenetic pathways, facilitating the identification of potential therapeutic interventions.
The authors note the current MONTE workflow can also be tailored to include or exclude enrichments based on the specific biological questions being addressed such as antibody-based phosphotyrosine peptide enrichment, further increasing the flexibility of serial enrichment workflows. All data from this study is publicly available, here.