
Glycosylation is critical for a wide range of biological processes and is implicated in numerous diseases. Analysis of the glycoproteome has the potential to reveal a wealth of clinical insights, but the heterogeneity of glycosylation makes this analysis difficult. Because of the analytical challenges resulting from the inherent heterogeneity of glycosylation, analysis of the glycoproteome has generally lagged behind other omics fields. To deal with this, a rapid expansion of software tools is underway in this area. In a recent study published in Molecular & Cellular Proteomics, CPTAC researchers propose a new approach for identifying the glycan component of an N-linked glycopeptide and an associated glycan FDR (false delivery rate) estimation method with two major advances from the existing methods. Combined with MSFragger glyco search and quantitation tools in FragPipe, the addition of glycan composition assignment and FDR control gives researchers a more comprehensive pipeline for fast and accurate analysis of data.
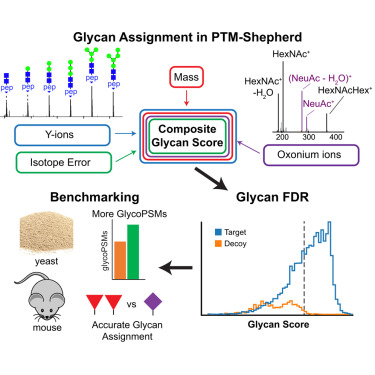 There are several combinations of monosaccharides that have equal or near-equal mass, making it easy for current FDR-based search engines to report the wrong glycan. Because of this, it remains common to manually validate search results to remove incorrect assignments, which serves to slow down larger-scale glycoproteomics studies. The majority of methods use a “glycan-first” approach, in which possible glycan candidates are first identified by matching the Y-ion series from the spectrum, determining the peptide mass, then searching for matching peptide fragment ions. This method is limited to data in which abundant Y-ions are produced, making it challenging to adapt for O-glycoproteomics. The CTPAC team proposes a “peptide-first” approach which relies on resolving the peptide portion of glycopeptide identification first and distinguishing between glycans that match that mass difference. A quote from the article summarizes the principle behind this approach:
There are several combinations of monosaccharides that have equal or near-equal mass, making it easy for current FDR-based search engines to report the wrong glycan. Because of this, it remains common to manually validate search results to remove incorrect assignments, which serves to slow down larger-scale glycoproteomics studies. The majority of methods use a “glycan-first” approach, in which possible glycan candidates are first identified by matching the Y-ion series from the spectrum, determining the peptide mass, then searching for matching peptide fragment ions. This method is limited to data in which abundant Y-ions are produced, making it challenging to adapt for O-glycoproteomics. The CTPAC team proposes a “peptide-first” approach which relies on resolving the peptide portion of glycopeptide identification first and distinguishing between glycans that match that mass difference. A quote from the article summarizes the principle behind this approach:
“By separating glycopeptide identification into peptide and glycan components and using the power of modern proteomics methods to solve the peptide part first, our method greatly simplifies glycan identification. Because the optimal fragmentation conditions typically differ for glycans and peptides, simplifying the identification problem in this way is key to confidently annotating more spectra and the glycopeptides they represent.”
The next step entails generating a composite glycan score from a variety of spectral evidence, including Y-ions, oxonium ions, and the observed mass and precursor isotope errors, rather than just Y-ions alone. This method has been implemented in the open-source tool PTM-Shepherd version1.2 and has been incorporated into the FragPipe graphical interface and pipeline to provide an accessible solution for glycoproteomics analyses.
As glycoproteomics moves to larger and more complex samples, confident assignment of glycan compositions is critical to move beyond manual validation of glycopeptide spectra. The separation of glycan assignment from peptide sequencing greatly simplifies the problem and enables the high performance of the researcher’s new proposed method. Team member Dr. Daniel Polasky elaborates on the importance of their work, writing:
“It is an important advance as there are many glycans with a similar mass that are hard to resolve, which is a big problem in the field. Distinguishing between similar mass glycans is a major challenge for data analysis methods in glycoproteomics but is essential to connecting biologically relevant changes in glycosylation to the proteins and sites at which it occurs. Combining FDR control of glycan compositions with the fast and sensitive search of MSFragger gives us the opportunity to do large-scale glycoproteomics with confidence.”
In addition, for the benefit of future research efforts in glycoproteomics, all raw data used can be found in public repositories noted in the Experimental Procedures section of the paper.