
Online on July 24th on Cell Systems, Clinical Proteomic Tumor Analysis Consortium (CPTAC) researchers shared the results of their first-ever, crowd-sourced NCI-CPTAC DREAM Proteogenomic Challenge. In this community-based, collaborative contest participants developed computational algorithms to predict protein abundance and phosphoprotein levels.
Focusing on the Issues
Changes in the genome drive carcinogenesis, but tumor progression 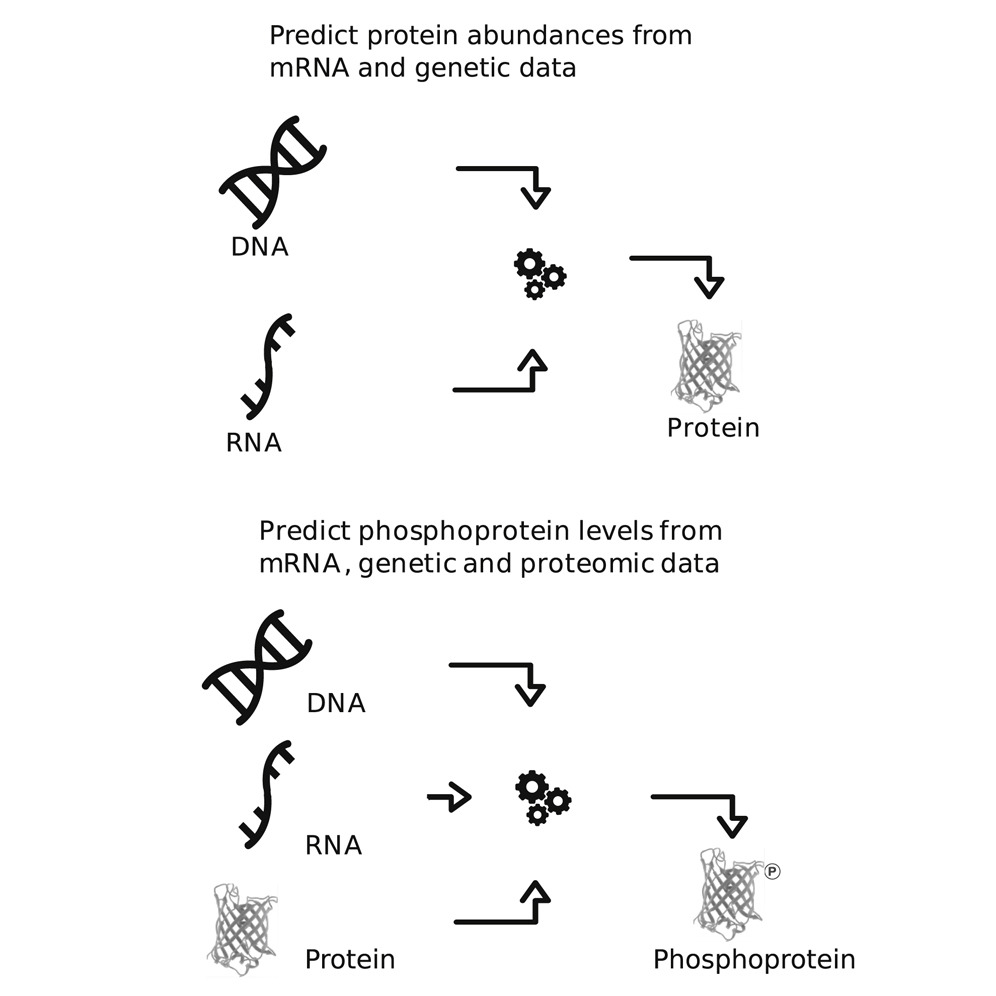 is largely driven by affected proteins making them the most used target of cancer treatment. Unfortunately, researchers are still in need of a cohesive understanding of how changes in the proteome effects the overall tumor biology, and in turn, how to more effectively treat them. At present, genomic and transcriptomic data of a given tumor is easier and less expensive to obtain, and less challenging to study than the human proteome.
is largely driven by affected proteins making them the most used target of cancer treatment. Unfortunately, researchers are still in need of a cohesive understanding of how changes in the proteome effects the overall tumor biology, and in turn, how to more effectively treat them. At present, genomic and transcriptomic data of a given tumor is easier and less expensive to obtain, and less challenging to study than the human proteome.
Recognizing these issues, Drs. Gustavo Stolovitzky (IBM Research), David Fenyo (New York University Grossman School of Medicine), Julio Saez-Rodriguez (European Molecular Biology Laboratory - European Bioinformatics Institute), and Henry Rodriguez (Director, Office of Cancer Clinical Proteomics Research-NCI) postulated whether there was a way to use readily available genomic and transcriptomic data to accurately predict protein and phosphorylation levels in cancer tissue. The idea was born to use the expertise of bioinformatists from across the globe in a crowd-sourced challenge to develop prediction algorithms which assess how much information about the differences in transcript and protein regulation is contained in the transcript quantities of other genes.
Finding Solutions
Challenge participants were tasked to develop various computational methods to accurately predict protein abundances from genomic and transcriptomic data, and also predict phosphoprotein levels from genomic, transcriptomic and proteomic data. Participants used genomic and transcriptomic data taken from two CPTAC cancer cohorts (breast and ovarian) to develop prediction algorithms. Participants were also able to use quantitative measurements of gene copy number, transcript, protein and phosphorylation levels for thousands of genes for two cancer cohorts and prior information from existing databases such as physicochemical properties and protein-protein interactions.
Once the wining team and top performers of the challenge were identified, a collective consensus algorithm was created. The best performing teams, including winners Drs. Yuanfang Guan with Hongyang Li, University of Michigan, mainly used transcript levels to predict protein levels and, for the phosphoproteomics prediction task, protein level was the most effective omics layer to predict phosphorylation levels.
The DREAM challenge outcome resulted in several key findings that elucidated transcriptional and translational control of the proteome and revealed factors influencing protein predictability, and biological pathways associated with the top predictor genes. For example, proteins not belonging to complexes were better predicted than those belonging to a protein complex. Also, metabolic and extracellular matrix pathway proteins were among the most predictable. However, the performance of even the best performing model was modest, suggesting that the levels of many proteins are strongly regulated through translational control and degradation.
The resulting consensus algorithm was applied to additional ovarian tumor data and showed that commonly predictive genes were more essential and predictive of patient survival for ovarian cancer. Also, using an ensemble model to infer phosphorylation levels significantly predicted overall patient survival for ovarian cancer and identified disease related mechanisms in the KEGG vascular muscle contraction pathway better than the corresponding protein level, illustrating the value of the methods produced in the challenge.
Understanding the extent protein levels can be predicted based on mRNA and genomic data alone provides researchers with models to understand protein-mRNA relationships and derive meaningful biological insights into cancer. Models from the DREAM Proteogenomic Challenge winners, and the DREAM challenge predications have been made publicly available for use and further development to the bioinformatic and cancer research community. Such large scale proteogenomic characterization of tumor samples could help us better understand cancer biology and identify novel biomarkers to predict survival and for patient stratification.
This study is a part of a series of studies supported by the NCI-CPTAC grants U24 CA210972 and U24 CA210993.