
In recent years, proteomic technologies have emerged as invaluable 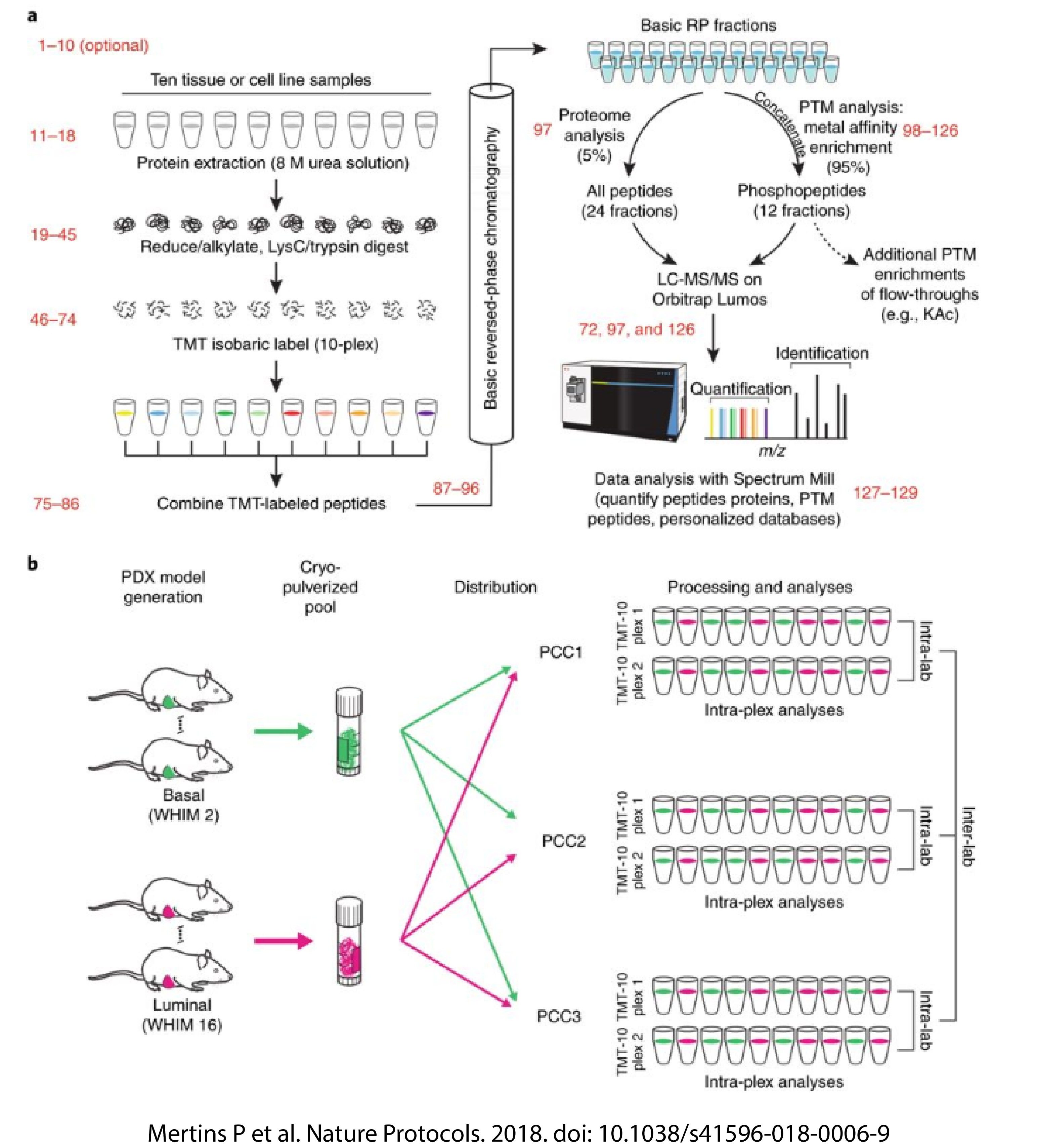 tools in cancer research. Next-generation mass spectrometry (NGMS) is being used to study cancer biology, while providing the cancer research community with a growing body of biological knowledge that may lead to more effective drug design, better patient treatment options, and more accurate prognoses.
tools in cancer research. Next-generation mass spectrometry (NGMS) is being used to study cancer biology, while providing the cancer research community with a growing body of biological knowledge that may lead to more effective drug design, better patient treatment options, and more accurate prognoses.
The National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC) has recently demonstrated that integrating deep, whole proteome data (protein abundance, phosphoproteomics) and genomics data from human tumor samples leads to a deeper understanding of tumor biology, anticipated to support the long-term goals of precision medicine. While analysis of these data highlights the strengths of mass spectrometry (MS)-based proteomics, achieving deep coverage of both the proteome and phosphoproteome with reasonable sample throughput and reproducibility presents as a challenge.
In a study recently published in Nature Protocols, CPTAC investigators optimized the workflow for deep, whole proteome and phosphoproteome profiling by implementing tandem mass tag (TMT) 10-plex isobaric labeling quantification. The optimized workflow increases throughput by three-fold with similar proteome coverage and high intra- and interlaboratory reproducibility quantification than previous protocols, while ensuring rigor & reproducibility. The protocol was developed using patient-derived breast cancer xenograft tumor models, and can also be applied to mammalian cell culture samples. The high quality, depth and reproducibility of data within and across laboratories will enable proteogenomic data integration and NGMS-based proteomics analyses across tumor types, yielding new biological insights and hypothesis-driven research questions. For more information regarding these improvements to the standardize workflows, please visit the CPTAC Data Portal.