
Genetic heterogeneity amongst leukemic cells is a major contributor to low survival rates and poor clinical outcomes for patients with acute myeloid leukemia (AML). This diversity drives complex signaling pathways at the protein level which necessitate individualized treatment protocols for each patient. In order to improve patient outcomes, ex vivo drug sensitivity assays to predict patient-specific drug responses are being researched. One such project, recently published in Clinical Proteomics, was designed by Clinical Proteomic Tumor Analysis Consortium (CPTAC) researchers to address an absence of direct information about proteomic signatures of drug response and demonstrates strong promise for proteomics-based patient stratification to assess drug sensitivity in AML.
This project leverages the rigorous pre-clinical drug testing and genomic profiling done by the Beat AML program with patient-derived proteomic and phosphoproteomic measurements to determine the potential for protein-level data to produce robust molecular biomarkers of drug response. Utilizing a proteomic dataset of 38 patients, the team evaluated drug sensitivity of 26 drugs targeting the FLT3, RAS/MEK, and other signaling pathways dysregulated in AML patients. The team endeavored to determine how well baseline proteomic and phosphoproteomic measurements can predict drug response compared to genomic or transcriptomic measurements. Study leader Dr. Karin Rodland spoke to their motivation for this investigation, saying “at present, precision oncology is using DNA mutations and RNA transcription data to predict tumor behavior and drug targets. We wanted to add protein and phosphoprotein data both alone/ in combination into the mix.” The team’s findings suggest that protein abundance alone and the combination of protein and RNA data are much stronger predictors of drug sensitivity than DNA alone (see below). As mentioned in their publication, robust performance of protein-based models compared to transcriptomic-based models opens up the possibility of developing antibody-based, CLIA-eligible assays for the rapid assessment of likely therapeutic targets at the time of biopsy, without the need for DNA or RNA sequencing.
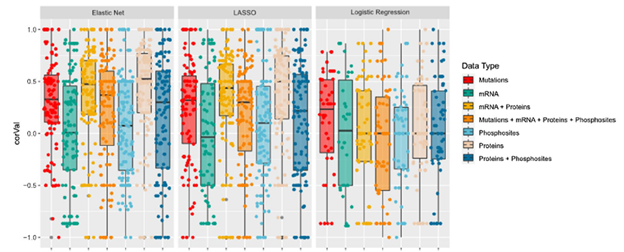
Figure 2B from the publication. This figure highlights the performance of multiple data types as predictors for drug response.
The group further examined top performing linear models to determine if they aligned with the known mechanism of action of their respective drugs and also experimentally validated these signatures in cell culture models of AML. In their publication, the group emphasizes the need for more interpretable models for drug response as regression approaches can fail to capture the entire cellular context contributing to AML pathogenesis and drug resistance. Mapping selected proteins using interaction networks could provide a better understanding of what causes drug resistance in some patients. Karin Rodland went on to comment that, “there has been a paradigm shift to look at not just behavior of tumor cells but what other cells like fibroblasts and immune cells are doing to contribute to the survival of the tumor. The whole molecular/cellular context is vital to our understanding of immune response.”
In summary, this study presents a strong argument for the effectiveness of proteomic and phosphoproteomic data as accurate predictors of drug response and establishes an effective workflow for the future analysis of integrated omics data. Looking forward, the team intends to expand their analysis using a larger patient cohort to continue investigating clinically relevant protein signatures which can be validated based on patient response data.