
Almost 18 months ago, CPTAC researchers from the Integrative Omics Group at Pacific Northwest National Laboratory (PNNL), developed a stream-lined technique to increase the sensitivity of peptide phospho-group identification during mass spectral analysis. The solution, called the ‘boosting to amplify signal with isobaric labeling’ (BASIL), was developed to ‘see’ small spectrometric signals (typically from post-translational modifications) that are hard to detect with conventional tandem mass spectrometry (MS/MS) in a small quantity of cells.
Well, they’re back again with an improved BASIL (iBASIL) 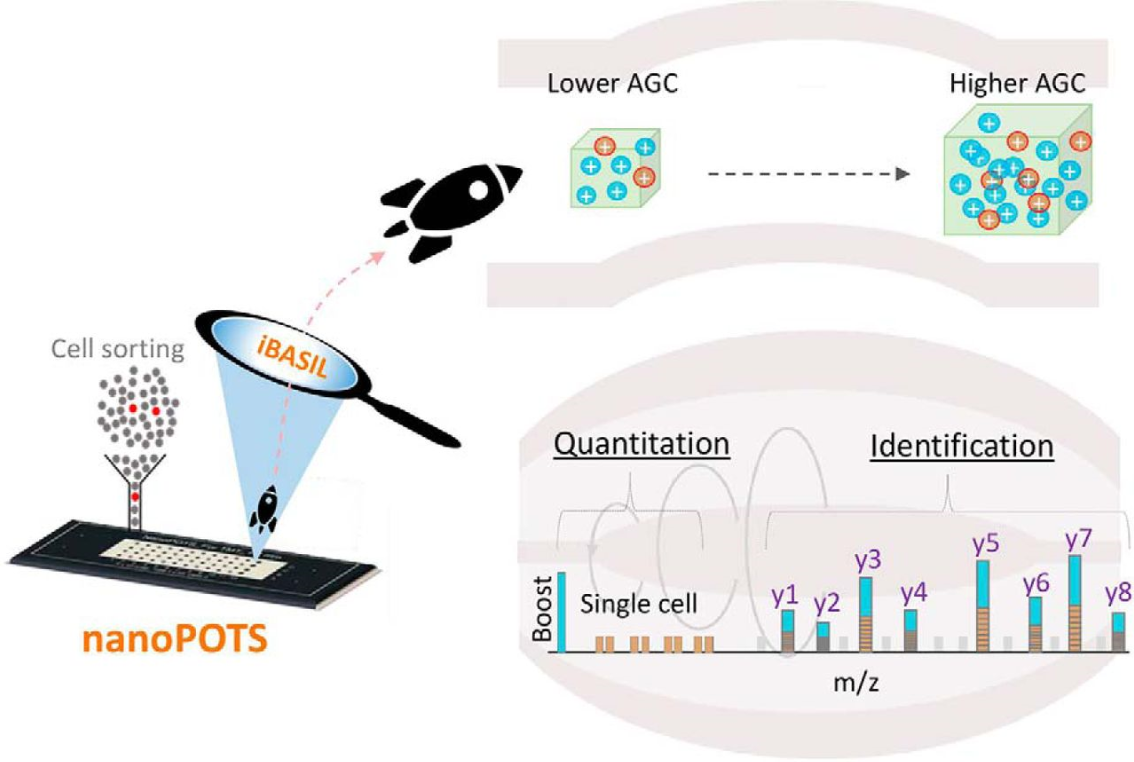 strategy that allows for the same increased peptide sensitivity – but on a single-cell level! So, what’s different? Dr. Chia-Feng Tsai, post-doctoral associate at PNNL and recently featured up-and-coming junior researcher in the Office of Cancer Clinical Proteomics Research Investigator Spotlight Series, and colleagues focused on two areas to improve sensitivity: automatic gain control and ion injection time.
strategy that allows for the same increased peptide sensitivity – but on a single-cell level! So, what’s different? Dr. Chia-Feng Tsai, post-doctoral associate at PNNL and recently featured up-and-coming junior researcher in the Office of Cancer Clinical Proteomics Research Investigator Spotlight Series, and colleagues focused on two areas to improve sensitivity: automatic gain control and ion injection time.
Let’s dive into that a little deeper. First, the team used their nanodroplet Processing in One Pot for Trace Samples (nanoPOTS) method to get the most efficient peptide yield for very small numbers of cells – even single cells. nanaoPots not only extracts proteins but digests them into peptides for mass spectrometral analysis. Then came the real magic. The team evaluated the boosting-to-sample ratios to reduce its effect on signal stabilities and signal-to-noise ratios of lower abundant proteins. Next, researchers saw that when using higher boosting ratios (for better sensitivity), instrument settings used in typical bulk analysis was not sufficient for effective detection of proteins in single-cell samples. To counter this, a significant increase in automatic gain control (e.g. 10X increase) was used, resulting in considerably improved reproducibility and lower missing values; ion injection timing was also lengthened significantly, allowing for an increase in the number of quantifiable peptides.
In their first BASIL article, Tsai and team tested feasibility on hard-to-obtain, small populations of human pancreatic islet cells. In this study, the improved method was demonstrated using acute myeloid leukemia (AML) single cells from 3 different AML cell lines. Researchers saw an increase of quantifiable peptides by 11-fold and proteins by almost 2-fold when applying the iBASIL strategy. Additionally, a greater than 4-fold increase of TMT signal in sample channels resulted in better quantitation and improved separation and clustering of the different AML cells. Coupling nanoPOTS with iBASIL enabled an unprecedented identification of ~2500 proteins and precise quantification of ~1500 proteins in the analysis of the AML single cells and revealed functionally distinct differences in their proteomes.
When it comes to single-cell analysis using the isobaric labeling-based boosting strategy, there are always trade-offs between achievable proteome coverage and quality of quantification. The iBASIL methodology demonstrates a way to look at single cells with the coverage and accuracy that rivals typical bulk analysis methods, paving the way for research solutions to important biological questions like tumor heterogeneity.