
Since its first mention in the scientific literature in 2004 by Jaffe et al1, the term proteogenomics has been shrouded in mystery and thick technical language. It is a complex idea, but one that is gaining traction as its value to improve our understanding of cancer development and potentially guide novel treatment strategies is being uncovered.
In the early 1990’s, advances in genomic sequencing technologies fueled the start of the Human Genome Project, opening-up a new world of information that was revolutionary to scientists learning about how the body works. In 2005, the focus of sequencing efforts turned to fully decoding the cancer genome. However, researchers have found that with all its invaluable insight into cancer biology, genomics and transcriptomics can’t answer all of our questions about cancer treatment.
Why is this? Simply put, genomics is a part of the ‘central dogma’, where DNA is the ‘input code’, RNA is the ‘translator’, and proteins are the ‘business end’ of everything that happens in our cells. Even after proteins are made, they are modified to turn on and off, to ‘see’ and ‘be seen’, and to be shuttled to where they need to be. It is for this reason that the vast majority of therapeutic drugs are targeted towards the regulation of protein functions.
Is it possible to make 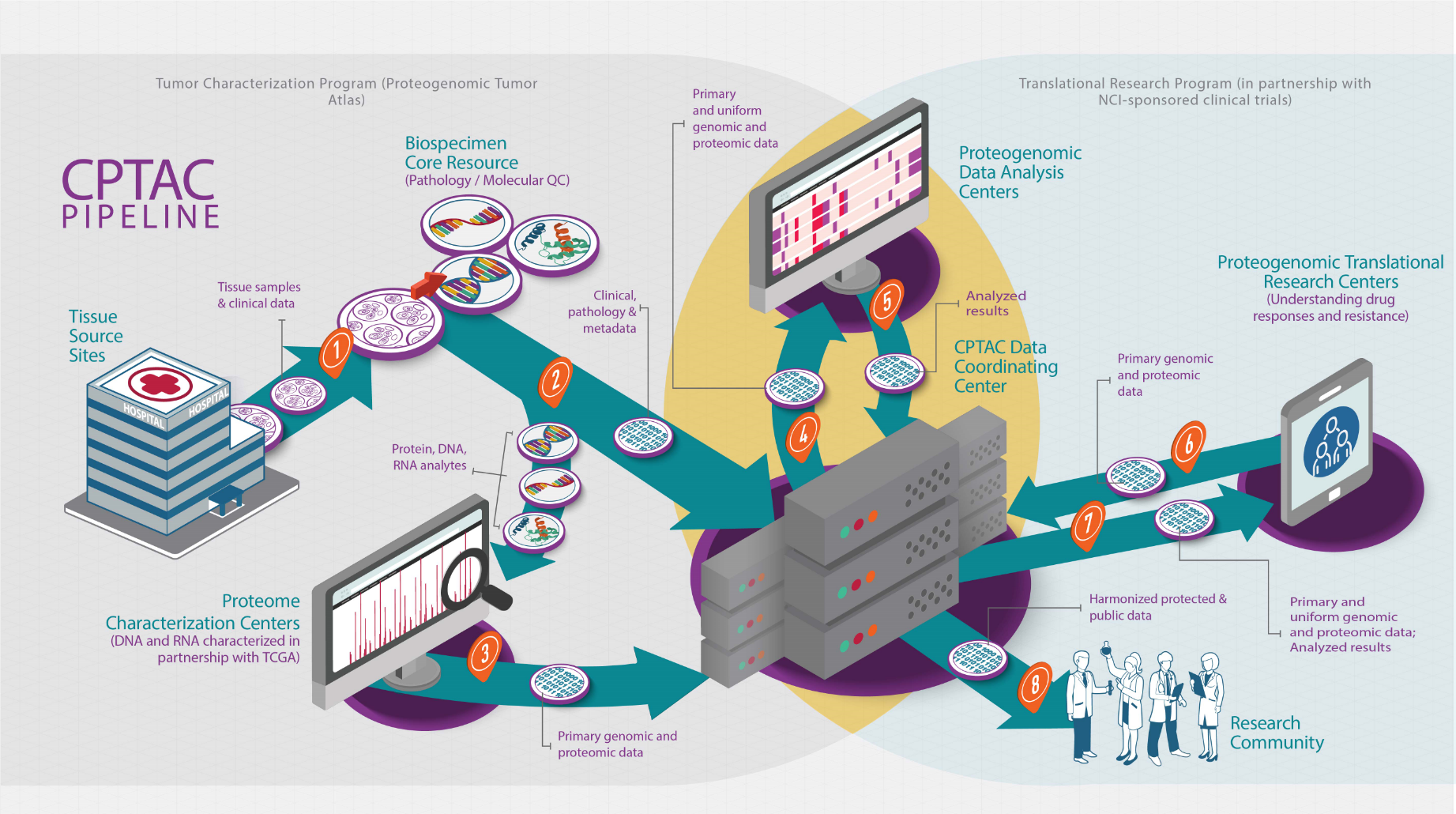 effective drug treatments without examining both the genomics and proteomics of a disease like cancer? Well, not all patients selected to receive targeted therapies based on genomic data experience lasting improvement of their disease, while others respond favorably. It is this baffling inconsistency that proteogenomics aims to elucidate.
effective drug treatments without examining both the genomics and proteomics of a disease like cancer? Well, not all patients selected to receive targeted therapies based on genomic data experience lasting improvement of their disease, while others respond favorably. It is this baffling inconsistency that proteogenomics aims to elucidate.
So what is proteogenomics? NCI’s Dictionary of Cancer Terms describes it as “the study of how information about the DNA in a cell or organism relates to the proteins made by that cell or organism. This includes understanding how genes control when proteins get made and what changes occur to proteins after they are made that may switch them on and off. Proteogenomics may help researchers learn more about which proteins are involved in certain diseases, such as cancer, and may also be used to help develop new drugs that block these proteins.“
Understanding the importance of this new field, the National Cancer Institute created the Clinical Proteomic Tumor Analysis Consortium (CPTAC) program to address the gaps in cancer research and standardize the implementation of proteomics as it relates to cancer.
CPTAC systematically identifies proteins that originate from alterations in cancer genomes (and associated biological processes) in order to understand the molecular basis of cancer and accelerate the application of those findings into the clinic. Proteogenomics enhances our understanding of the cancer genome biology by helping prioritize genomic alterations, enhancing the understanding of pathogenesis through proteomic subtyping, illuminating dynamic alterations among posttranslational modifications responsible for the dysregulation of cancer signaling networks and pathways, and improving the understanding of drug response and resistance to therapies.
Let’s unpack how CPTAC creates this cancer proteogenomics resource. The CPTAC tumor characterization pipeline starts when a cancer patient consents to participate in a study by donating leftover tumor tissue to the CPTAC proteogenomics program to further cancer research. The tumor sample is then submitted to a CPTAC Tissue Source Site (TSS) that collects tumors from institutions around the world. The TSS performs some initial examinations and ships the tissue to the CPTAC Biospecimen Core Resource (BCR), where it is further processed, and DNA and RNA are isolated.
The tissue and its components then move to the next step in the pipeline - a Proteome Characterization Center (PCC), and its DNA and RNA are sent to Genomic Characterization Centers (GCC) for sequencing. To examine the proteins, PCC experts use a mass spectrometer (MS), a specialized instrument that breaks up proteins into smaller pieces (called peptides), which are then divided by size with the smallest peptides travelling first through the MS chambers. As the peptides are sprayed into the main chamber of the MS they are bombarded with lasers, like in laser tag (ionized), to knock electrons off and make them positively charged. They then fly along the MS and hit a detector at specific places according to how heavy they are and the amount of charge they have. They result is a spectrum that is a unique fingerprint for each peptide.
Once the peptides have been identified based on their spectra, and together with the genomic and transcriptomic sequencing data, all information is sent to a CPTAC Proteogenomic Data Analysis Center (PGDAC) where investigators pour over it. Using computer algorithms, computational strategies and database libraries, they stitch the information for each protein in the central dogma back together, making the myriad of data comprehensible.
The final conclusions from integrative analysis of all the data is also shared with CPTAC Proteogenomic Translational Research Centers (PTRCs). They figure out which proteins might make interesting markers and targets. What makes a protein interesting? Knowing whether protein targets are a part of a biological pathway that effects a tumor’s ability to flourish makes a protein of particular interest to researchers, as well as how these targets have been affected – whether they are turned off or on, etc. Additionally, these targets can help explain which patients will benefit from a specific therapy. All this information helps researchers develop effective ways to fight cancer.
To make sure cancer researchers have the most complete information available to find new or improved cancer drug targets, CPTAC makes its data publicly available for anyone to use through its CPTAC Data Portal, the Genomics Data Commons, and The Cancer Imaging Archive. Presently, the CPTAC program has complete datasets (including genomics and imaging) publicly available for:
- uterine corpus endometrial carcinoma (UCEC),
- clear cell renal cell carcinoma (ccRCC), and
- lung adenocarcinoma (LUAD).
This is an exciting time for cancer research, and the CPTAC program is leading the way to discovering new cancer therapies. Sign up here to get email alerts about upcoming proteogenomic data releases.
1. Jaffe JD, Berg HC, Church GM. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004;4:59-77.