
Using a single-needle biopsy and new technology for 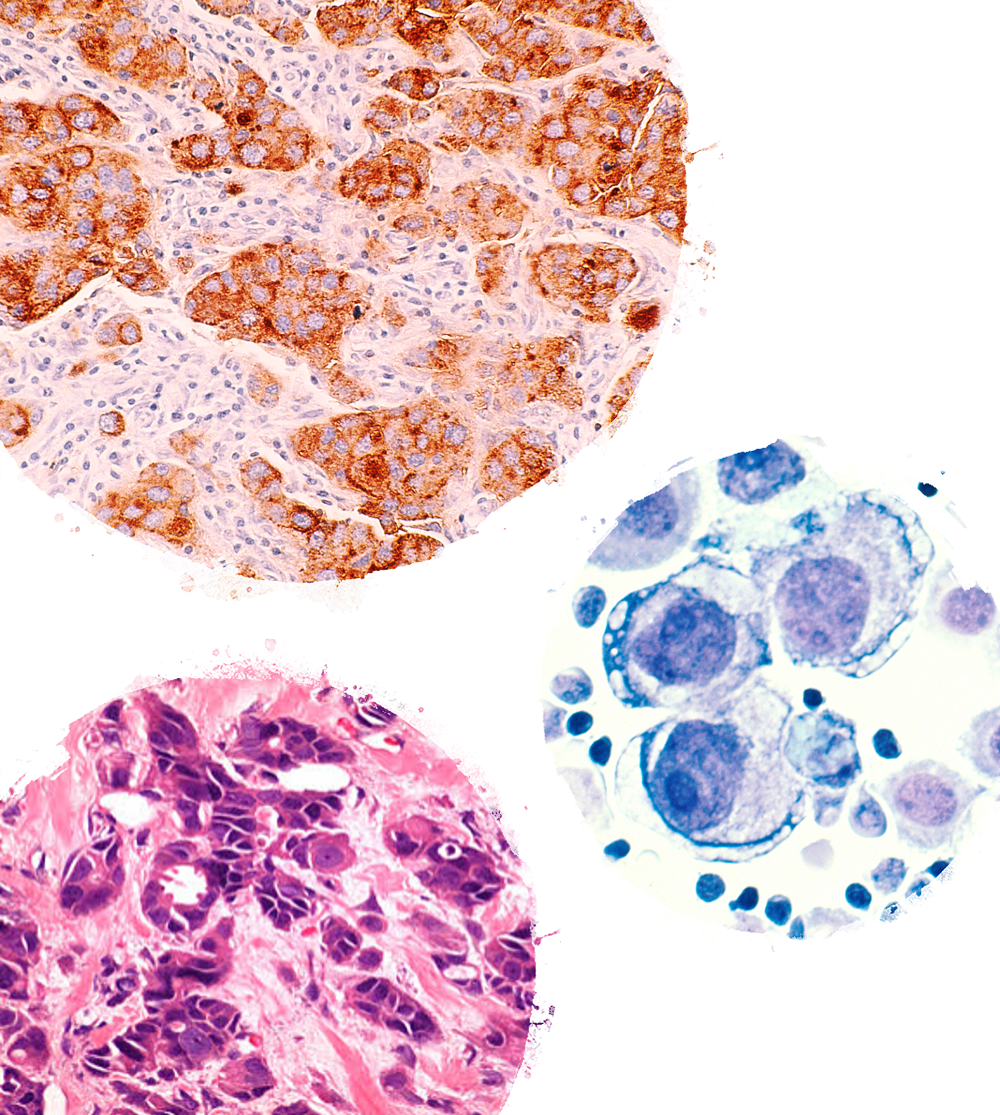 tumor diagnosis developed by Baylor College of Medicine and the Broad Institute of MIT and Harvard, researchers have been able to provide a more detailed and wider window into cancer biology, tumor type and the mechanisms of response and resistance to therapy than with conventional approaches.
tumor diagnosis developed by Baylor College of Medicine and the Broad Institute of MIT and Harvard, researchers have been able to provide a more detailed and wider window into cancer biology, tumor type and the mechanisms of response and resistance to therapy than with conventional approaches.
This new-method analyzes the combination of tumor genetic material (genomics) with deep protein and phosphoprotein characterization (proteomics) using a single-needle core biopsy from a patient’s tumor. This type of proteogenomic analysis had only been possible before with much larger tissue samples taken at surgery and shows promise for future use in clinical application.
The study outlining this new micro-scale technology appears in the journal Nature Communications.
The need for new technology
“Patients die from cancer because, at a sufficiently fundamental level, we have not been able to work out what kind of cancer we are treating,” said co-corresponding author Dr. Matthew Ellis, professor and director of the Lester and Sue Smith Breast Center, McNair scholar, and associate director of Precision Medicine at the Dan L Duncan Comprehensive Cancer Center at Baylor. “The analysis of proteogenomics data, which combines information on tens of thousands of proteins and genes together using a system developed by the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium investigators, provides much more complete details about what is going on in each tumor. However, the application of proteogenomics to both scientific research and cancer diagnosis has been limited by the size of the tissue sample required.”
“Typically, patient tumor biopsies are about one-fifth the size of what is needed for proteogenomics analyses, so we invested considerable effort to identify methods to successfully ‘microscale’ the process to match the smaller amount of material obtained in the routine diagnostic setting and, importantly, not sacrifice depth of coverage of the proteome and phosphoproteome,” said Dr. Steven Carr, co-corresponding author and senior director of proteomics and institute scientist at the Broad Institute.
The published study describes the methods developed that enable detailed analyses of high-quality DNA, RNA and proteins from a single core needle biopsy.
“Importantly, our new methodology includes the analysis of phosphoproteins, which refers to proteins that are activated by the addition of phosphate chemical groups,” said Ellis, who also is an investigator in the NCI-CPTAC. “For some cancers, such as ERBB2+ (HER2+) breast cancer, the ability to measure these modifications is critical because they are what drives disease.”
Testing the new technology
To test the new technology, Carr, Ellis and their colleagues applied the methods to a pilot study designed to evaluate the feasibility of proteogenomic profiling before and 48 to 72 hours after initiating ErbB2 targeting chemotherapy. They expected to gain insights into the variability of outcomes after treatment by assessing the ability of ErbB2 antibodies to inhibit the drug target.
“For the first time, we were able to detect statistically significant reduction of ERBB2 protein phosphorylation after treatment in patients that responded to treatment. We did not see a reduction in this protein for those who did not respond to treatment,” Ellis said. “In patients that did not respond to treatment, our deep-scale data analyses suggested diverse resistance mechanisms to ERBB2-directed therapeutics that could be addressed with alternative approaches to the ones the patient actually received.”
The test of the micro-scaled technology provided large amounts of data from the tumors, revealing fundamental insights into the diverse elements that drive tumor responses, including those from the tumor immune microenvironment. The test served as proof of principle that these technologies have promise for precision medicine, meaning it can be used to examine individual tumors and find precise treatment plans for each one.
“However, before we bring the new technologies to the clinic, it is necessary to apply them to a larger number of tumor samples to confirm their diagnostic value,” Ellis said.
“Under the auspices of the NCI-CPTAC, we will expand studies into larger groups of patients undergoing treatment for breast cancer in the context of clinical trials,” said Carr, a co-principal investigator with Ellis in the NCI-CPTAC.
A key challenge will be access to a large number of high-quality, preserved clinical samples. The feasibility of learning insightful biology as demonstrated in this paper should provide an incentive to clinicians and clinical trial teams to collect and store biopsy material for large-scale prospective studies and for understanding the biology of drugs in patient samples.
“The ability to study signaling pathways in patients is unparalleled,” said first and co-corresponding author Dr. Shankha Satpathy, a research scientist in the Carr lab at the Broad.
“Moving forward, our plan is to develop a laboratory at Baylor to conduct these types of diagnosis using micro-scaled proteogenomic technologies in order to provide a better diagnostic platform for our patients,” Ellis said.
“I am particularly excited by the potential to move from the current discovery paradigm that requires considerable time to generate the results to a targeted approach that will enable rapid profiling of large lists of proteins we illustrate in our proof of principle paper,” said Carr. “But the first step is to define what should be measured that is of biological significance and clinically actionable, as opposed to just what can be measured. That is the most important contribution that our technology is making.”
Other contributors to this work include Eric J. Jaehnig, Karsten Krug, Beom-Jun Kim, Alexander B. Saltzman, Doug W. Chan, Kimberly R. Holloway, Meenakshi Anurag, Chen Huang, Purba Singh, Ari Gao, Noel Namai, Yongchao Dou, Bo Wen , Suhas Vasaikar , David Mutch, Mark A. Watson, Cynthia Ma, Foluso O. Ademuyiwa, Mothaffar Rimawi, Rachel Schiff, Jeremy Hoog, Samuel Jacobs, Anna Malovannaya , Terry Hyslop, Karl C. Clauser, D.R. Mani, Charles Perou , George Miles, Bing Zhang and Michael A. Gillette. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine; Broad Institute of Harvard and Massachusetts Institute of Technology; Siteman Comprehensive Cancer Center and Washington University School of Medicine; NSABP Foundation; Duke University Medical Center, Durham; University of North Carolina at Chapel Hill and Massachusetts General Hospital, Boston.
This work was supported by the U.S. National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) and grants from the NIH/NCI
U24-CA210986, NIH/NCI U01 CA214125, NIH/NCI U24CA210979, NIH/NCI U24 CA210954, NIH/NCI U10 CA180860 and NIH/NCI U54CA233223. Tissue acquisition was partly supported by a Breast Cancer Research Foundation (BCRF) grant and was also supported by Cancer Prevention & Research Institutes of Texas Scholar (CPRIT) established investigator recruitment award CPRIT RR140033. Further support was provided by a Susan G. Komen Scholar and McNair Medical Foundation Fellowship and a Cancer Prevention & Research Institutes of Texas Scholarship in Cancer Research (CPRIT RR160027), a Gift from the Ralph and Lisa Eads Family and a McNair Medical Institute Scholarship. Additional support was provided by the NCI Cancer Center Support Grant #P30 CA091842 and the ICTS is funded by the National Institutes of Health’s NCATS Clinical and Translational Science Award (CTSA) program grant #UL1 TR002345.