
Large multi-omics datasets are becoming increasingly 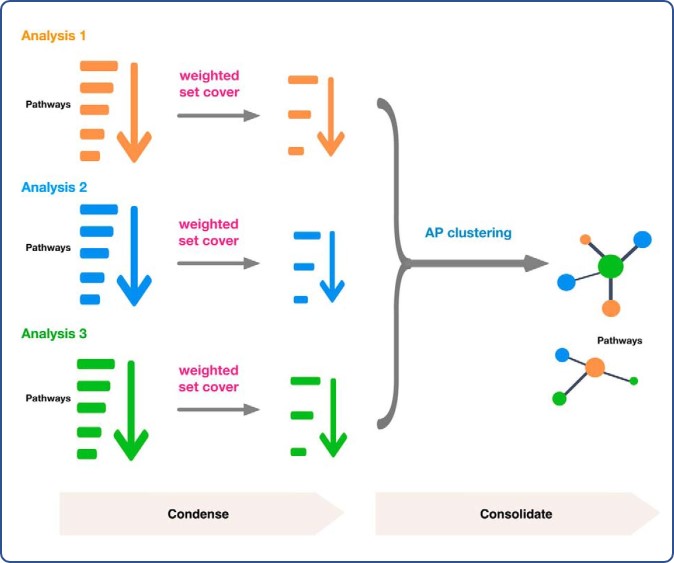 popular for studying biological systems including the identification of biological pathways, or more broadly defined gene sets, that are associated with certain biological or clinical features of interest. The consolidation of cancer proteogenomic data (which includes DNA, RNA, proteins, phosphorylated proteins, etc.) to identify biological pathways associated with different cancer genotypes or phenotypes within or across different cancer types becomes the basis for better personalized medicine, fostering development of new therapies to overcome resistance.
popular for studying biological systems including the identification of biological pathways, or more broadly defined gene sets, that are associated with certain biological or clinical features of interest. The consolidation of cancer proteogenomic data (which includes DNA, RNA, proteins, phosphorylated proteins, etc.) to identify biological pathways associated with different cancer genotypes or phenotypes within or across different cancer types becomes the basis for better personalized medicine, fostering development of new therapies to overcome resistance.
The analytical process of integrating huge volumes of genomic and proteomic datasets from different omics platforms and distilling them into accurate disease pathways is fraught with complexity. CPTAC investigators Drs. Bing Zhang, Sara Savage and Zhiao Shi have found a way to reduce gene set redundancy problems in the same, or different, databases such as GO, KEGG and clusterTools to allow for clearer pathway associations and conclusions. In their paper published in Molecular & Cellular Proteomics, Dr. Savage lays out how their team used both weighted set cover and affinity propagation algorithms integrated into an R-platform to create the Sumer program to define gene sets for use in identifying biological pathways involved in disease and allowing for pan-disease comparisons.
Briefly, in a two-step process, researchers first used weighted set cover algorithms applied to multiomic sources to reduce gene annotation redundancy in individual enrichment analysis. This was followed by use of affinity propagation algorithms to cluster the results from multiple enrichment analyses. Affinity propagation allows not only grouping of similar sets of affected genes that become apparent through multiomic analysis, but also identifying the most representative gene set for each group. Rolling these algorithms into the Sumer program, the researchers were able to integrate enrichment analyses from multi-omics data into a single study and further, to integrate the results from multiple studies.
As a proof of concept, CPTAC investigators applied this technique to several multi-omics, pan-cancer studies to show cancer-specific features in biological pathways. Examining the expression of certain DNA-damage response pathway-associated genes, for instance, they were able to demonstrate a negative association with survival in lung adenocarcinoma and kidney cancer, whereas a similar set of genes displayed an association with survival in colorectal and endometrial cancers.
Dr. Savage and team have demonstrated a technique to extract accurate and relevant information from a large amount of proteogenomic data. CPTAC represents a team of leaders in the field of cancer proteogenomics, making strides in cancer research to accelerate precision oncology.