
In a new study published in Analytical Chemistry, we see another example of ingenuity by NCI’s Office of Cancer Clinical Proteomic Research (OCCPR) Clinical Proteomic Tumor Analysis Consortium (CPTAC) scientists. In an effort to optimize analytical output and conserve hard-to-obtain biospecimen samples, CPTAC researchers out of John Hopkins School of Medicine developed a new integrated workflow for multiplex analysis of global, glyco-, and phospho-proteomics.
Why is this needed? In tumor biospecimens 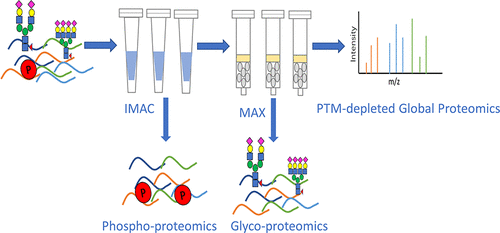 post-translational modifications (PTMs) of proteins, such as glycosylation and phosphorylation, are often difficult to detect because they make up a small subset of the overall proteomic signal in a sample. When researchers need to carry out several distinct PTM analyses in addition to global proteomic analysis, this small amount of tissue collected from patient tumors are often divided into several aliquots, each aliquot being specifically enriched for the target PTM to be analyzed by LC-MS/MS. Subsequently, the amount of available tissue can be the limiting factor of what types of analyses are done.
post-translational modifications (PTMs) of proteins, such as glycosylation and phosphorylation, are often difficult to detect because they make up a small subset of the overall proteomic signal in a sample. When researchers need to carry out several distinct PTM analyses in addition to global proteomic analysis, this small amount of tissue collected from patient tumors are often divided into several aliquots, each aliquot being specifically enriched for the target PTM to be analyzed by LC-MS/MS. Subsequently, the amount of available tissue can be the limiting factor of what types of analyses are done.
Further, in conventional global proteomic analyses, some MS spectra remain unassigned. Recent studies have suggested that these unassigned spectra might be from unidentified PTMs or low abundant proteins. Therefore, the development of an efficient, integrative method that allows for several types of proteomic analyses on a single aliquot of tumor tissue would be ideal.
Using breast cancer patient-derived xenograft (PDX) tumor samples, CPTAC investigators explored the use of immobilized metal-ion affinity chromatography (IMAC) and mixed ion exchange (MAX) enrichment steps for phosphoproteomics and glycoproteomics respectively, prior to global proteomics on a single sample.
Briefly, this new workflow begins with IMAC enrichment of the initial sample for phosphoproteomic analysis using mass spectrometry, followed by removal of phosphate groups using phosphatases. IMAC enrichment is simple to do and allows for high labeling specificity, and reproducibility. The same sample was then enriched for glycopeptide enrichment using MAX for glycoproteomic analysis, followed by removal of glycolic groups using glycosidase. MAX enrichment was chosen because of its superior performance on glycopeptides. Finally, the PTM-depleted sample was analyzed a for global protein analysis.
Using this method researchers were not only able to demonstrate an increase in peptide identification and associated proteins for the global proteome, but able to multiplex these analyses on the same sample for protein PTM analyses.
Among the many advantages of this new integrated workflow, CPTAC scientists were able to demonstrate successful simultaneous global, phospho-, and glyco-proteomic analyses within the same sample, while discovering even more unique PTMs and proteins! In one word - multiproteomics.